Development of the Embryo
The union of individual male and female gametes represents an orderly and highly regulated sequence of events collectively termed fertilization.14 Fertilization involves maturation of spermatozoa15,16,17 by capacitation, and their movement through the cumulus of the ovum,18 although human sperm need not undergo capacitation to traverse the cumulus.16 Sperm binding to zona pellucida proteins (ZP1-4), primarily ZP3,19,20 precedes the acrosome reaction21 that is essential for sperm fusion with the oocyte membrane.22 After fusion of the egg and sperm membranes, release of cortical granules in the oocyte results in conformational changes in ZP2 and ZP3 that render the ovum impenetrable to additional sperm.16
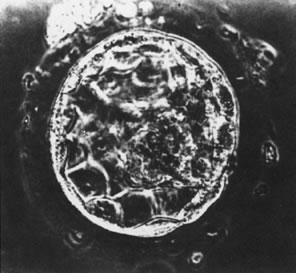
At the 8-cell stage, blastomeres begin to form gap and tight junctions,23,24 a process that initiates segregation of inner cells from outer cells and marks the onset of embryo differentiation.25 After additional cell divisions, the embryo (now termed a morula) enters the uterus, approximately 4 days after ovulation in both humans and nonhuman primates.26 After formation of the blastocoele, cells of the blastocyst differentiate into an inner cell mass destined to form the fetus and an outer mass of cells destined to become the placenta (Fig. 1).27 This process appears to be modulated by cell adhesion molecules, since E-cadherin-null embryos fail to form a trophectoderm.28 The developing blastocyst continues to grow and essentially floats in the uterine cavity for an additional 2 to 3 days during which time the blastocyst (embryo) hatches from the surrounding zona pellucida.29,30,31 Hatching appears to be essential for embryo-uterine contact and for subsequent implantation that occurs between 7 and 9 days after ovulation in humans and most nonhuman primates. Enders32,33 has concluded that prior to attachment to the uterine epithelium, mononucleated cytotrophoblast cells of the trophectoderm fuse to form a syncytiotrophoblast layer. The syncytiotrophoblast appears to initially interact with and adhere to the endometrium. Only after the embryo is totally embedded in the endometrium do cytotrophoblast cells begin to move from the trophoblastic shell to invade the uterus and the uterine vasculature.34,35,36
|
Preparation of the Uterus for Implantation
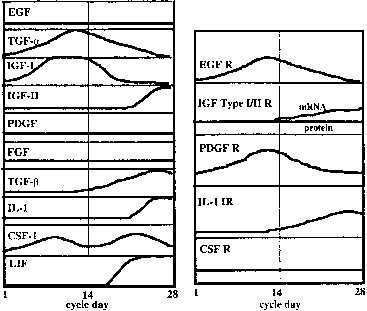
As the embryo (blastocyst) is developing and moving through the fallopian tubes into the uterus, the endometrium undergoes extensive differentiation ultimately to permit embryo attachment (i.e., implantation). In virtually all mammals, preparation of the uterus for implantation is regulated by the coordinated actions of estrogen and progesterone37 produced and secreted by the ovary-corpus luteum and perhaps by the developing embryo itself.38,39 Estrogen and progesterone act on the endometrium and myometrium directly through estrogen-progesterone receptor-mediated events, and indirectly by stimulation of various growth factors (e.g., EGF, TGF-B, insulin-like growth factor I (IGF-I) and IGF-II), proteins (e.g., placental protein 14, 24 kd protein, CA-125), and cytokines (e.g., IL-1, CSF-1) (Fig. 2).40 Uterine receptivity to a putative implantation signal is limited to a discrete period of time during the luteal phase of the menstrual cycle consistent with the concept of a window of implantation that has been advanced in several animal models and in the human.41,42,43 However, despite intense investigation, the molecular basis for the transient nature of uterine receptivity, estimated to span days 19/20 to 24 of the menstrual cycle,35,44,45 remains unknown. During this interval, and presumably under the influence of estrogen and progesterone, the uterine endometrium is thickened and highly secretory in nature and becomes rich in glycogen and lipids.37 Morphologic studies have shown that uterine receptivity is heralded by the formation of pinopods on the apical surface of endometrial epithelial cells, a process that appears to be regulated by progesterone.46 Pinopods may absorb fluid from the uterine cavity, thereby making the endometrium more accessible to the blastocyst. Changes in the composition of the uterine glycocalyx have also been observed during the peri-implantation period. For example, the levels of two transmembrane glycoproteins, mucin (MUC-1) and keratan sulfate, increase on the endometrial glandular surface during the early luteal phase,9,47,48 then decrease as the window for implantation opens.9,48 Because MUC-1 is a relatively large cell surface molecule, its down-regulation may unmask smaller molecules on the uterine surface, such as the cadherins and integrins, thereby mediating specific adhesion of the trophectoderm to the endometrium. Epithelial integrins have been proposed as markers for the window of implantation in the human.49,50 Endometrial integrins are expressed in an “on/off” pattern,51 and both they and their ligands are expressed in trophectoderm.52 Taken together, these observations support the hypothesis that integrins may be involved in early implantation events. The β 3 integrin subunit is expressed on the endometrial surface after day 19 of the human menstrual cycle, just when the endometrial window of implantation opens.53 The fact that this integrin is almost never expressed by epithelial cells, but is expressed by human trophectoderm,54 supports a role for these molecules in implantation. A similar distribution of endometrial integrins and extracellular matrix proteins has been described across the menstrual cycle and early pregnancy in the baboon.55 Human secretory phase endometrium also produces the glycoprotein leukemia-inhibiting factor(LIF).56,57 LIF has the capacity to inhibit embryonic stem cell differentiation in vitro,58 and LIF receptors are detected on human blastocysts.56,57 Moreover, Stewart and colleagues59 have demonstrated that LIF (production of which is regulated by estrogen and progesterone) is essential for implantation in the mouse since implantation of blastocysts did not occur in mutated mice lacking a functional LIF gene. A similar role for LIF in humans and nonhuman primates has not been confirmed.
|
In addition to expression of integrins, the human blastocyst/embryo secretes a number of factors at various times during its development that may be essential for pregnancy recognition or its normal progression. Almost immediately after fertilization, the embryo secretes platelet-activating factor (PAF), interleukins-1 and -6, and early pregnancy factor. Although inhibition of PAF activity in vivo prevented implantation in rodents,60 a similar role in primate pregnancy has not been demonstrated. By the 8-cell stage, the blastocyst apparently secretes a number of cytokines and growth factors including chorionic gonadotropin(hCG), long recognized as one embryonic factor that is essential for early pregnancy in primate species.31,61,62,63,64
Rescue of the Corpus Luteum by Human Chorionic Gonadotropin
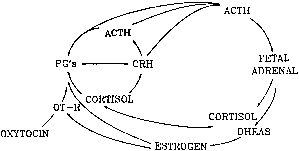
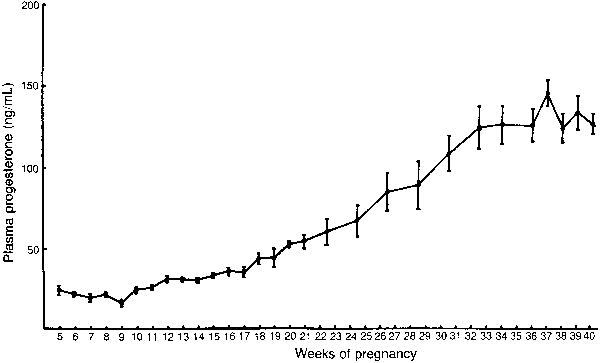
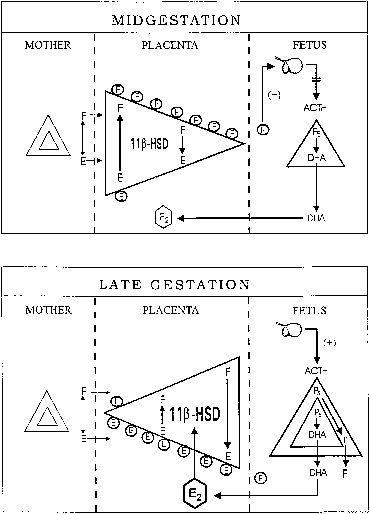
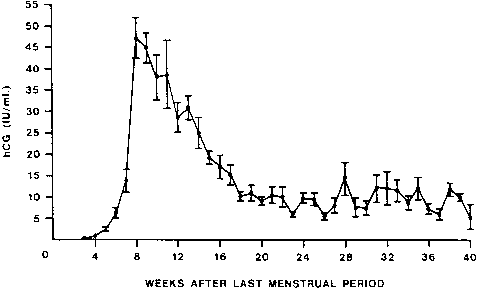
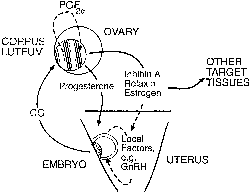
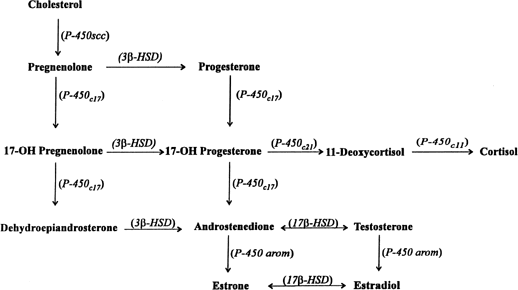
hCG is a glycosylated protein heterodimer composed of noncovalent bound α and β subunits.65 β-hCG is structurally similar to the β subunit of pituitary luteinizing hormone (LH), differing only in the terminal 28 amino acids. Whereas α subunit genes are expressed in the pituitary and placenta,66 the 6 β subunit genes located on chromosome 19q13.3 are expressed only in the placenta.67 The three-dimensional structure of hCG has also been determined68; both the highly glycosylated α and β subunits69 contain several disulfide linkages that form cysteine knot motifs.68,70 Deglycosylation studies indicate that the carbohydrate moieties of the α, but not the β, subunit are critical for activation of the hCG receptor and its associated G-protein coupled signaling systems.70 In human pregnancy, hCG levels in maternal blood are detectable approximately 3 days after implantation71 and rapidly rise thereafter with peak levels being achieved between weeks 8 and 12 of gestation (Fig. 3).72 Although in vitro studies suggest that hCG may be produced by hatched embryos prior to attachment, expression is generally assumed to occur after attachment.26,73 In the absence of hCG, the newly formed corpus luteum will invariably regress; production of estrogen and progesterone declines resulting in endometrial shedding (i.e., menses). The hCG produced by the syncytiotrophoblast binds to the LH/CG receptor in the corpus luteum to stimulate progesterone and estradiol synthesis, presumably by increasing low-density lipoprotein receptor expression and thus facilitating uptake of cholesterol substrate for steroidogenesis (Fig. 4).31 Immunohistochemical studies also suggest that hCG may act to increase the expression of rate limiting steroidogenic enzymes (e.g., 3β-hydroxysteroid [-HSD] and aromatase; Fig. 5).74 In some species, but perhaps not the human, rescue of the corpus luteum by hCG may also involve local inhibition of factors (e.g., prostaglandin) that promote luteal demise (e.g., luteolysis). hCG is also thought to stimulate corpus luteum relaxin production (see Fig. 4) and, as discussed later, appears to enhance testosterone production by the fetal testes that is required for differentiation of the internal duct system and external genitalia in males. The high levels of hCG secreted into the maternal circulation during early gestation apparently are sufficient to bind to the thyroid-stimulating hormone (TSH) receptor and thereby to increase maternal thyroid hormone production.75 Although our current understanding of the regulation of hCG production remains incomplete, it has been proposed that gonadotropin-releasing hormone (GnRH) produced by the placental cytotrophoblast may modulate hCG production by binding to GnRH receptors in the syncytiotrophoblast.5 Interestingly, human placental trophoblast produces a variety of other hypothalamic-/pituitary-like neuropeptides,5 including corticotropin-releasing hormone (CRH), proopiomelanocortin (POMC), neuropeptide Y, oxytocin, somatostatin, and thyrotropin-releasing hormone (TRH). Although the physiologic role of most of these factors remains to be defined, CRH may play a role in parturition,76,77 modulation of uterine blood flow,78 and regulation of maternal pituitary ACTH production.79,80 Finally, the trophoblast also produces several protein hormones (chorionic somatomammotropin [hCS] and growth hormone variant [GH-V]) and growth factors (IGF-I and IGF-II) that are considered essential to the regulation of maternal, and perhaps fetal, intermediary metabolism as well as growth and maturation of the placenta.81
|
|
|
Placental Development, Vascularization, and Intrauterine Endovascular Invasion
DECIDUALIZATION
Upon attachment of the embryo to the uterus, the uterine endometrial glycogen-containing stromal cells are rapidly transformed into large decidual cells that increase in size throughout the course of gestation. The human decidua that represents the maternal component of the placenta is composed primarily of the decidua basalis, underlying the site of implantation, the decidua capsularis that initially overlies the gestational sac but gradually disappears with advancing gestation, and the decidua vera that lines the remainder of the uterine cavity. In humans, as well as the baboon,82 the decidua produces and secretes a variety of factors that include the hormones relaxin and prolactin, the IGF-binding protein (IGF-BP-1), and a variety of other proteins (e.g., placental protein 14, also known as progesterone-associated endometrial protein [PEP], or glycodelin).
In the human, IGF-BP-1 is secreted by the stromal cells that surround the spiral arteries during the late luteal phase, whereas in the baboon, IGF-BP-1 is secreted by the endometrial glands in response to progesterone.83,84 IGF-BP-1 is one of several proteins that bind IGF-I and IGF-II and thereby regulate the ability of these growth factors to interact with their receptors. Ritvos and associates85 have suggested that decidual IGF-BP1 may act to control invasion and/or proliferation of trophoblast cells during implantation/placentation by sequestering the IGFs. IGF-BP-1 levels increase rapidly during early gestation in parallel with decidualization, then transiently decline before increasing again in late pregnancy.86,87 Pregnancy termination with the progesterone receptor antagonist RU-486 is associated with a marked decline in IGF-BP-1 levels, an observation suggesting that decidual IGF-BP-1 production is progesterone dependent.53 It has also been hypothesized that restructuring of the decidual cell cytoskeleton is essential for IGF-B1 gene expression.88
Glycodelin, synthesized by the uterine glandular epithelium, shares homology with the B-lactoglobulins and retinol-binding proteins89 and has been implicated as an immunosuppressive agent.90,91 Close temporal relationships in serum profiles suggest that glycodelin production is regulated by progesterone92,93 and/or relaxin.94 Serum glycodelin levels are elevated in the luteal phase of the menstrual cycle and markedly increase during the first trimester of pregnancy. In the baboon, although the pattern of uterine glycodelin mRNA and protein expression mimics that in the human, glycodelin production is regulated by hCG,95 perhaps by direct action on the endometrium.
Decidual prolactin production begins on day 22 of the idealized human menstrual cycle (approximately 8 days after ovulation)96 and prolactin mRNA and protein expression is observed in the epithelial cells of the deep basal glands of the baboon uterus during the late luteal phase. Decidual prolactin expression increases markedly with advancing gestation and is stimulated by progesterone in both the human97,98 and baboon.99 Decidual prolactin is apparently secreted into the amniotic fluid in humans100,101 and baboons,102 and levels increase with advancing gestation. Although the precise role(s) of decidual prolactin remains unclear, it has been proposed that the hormone may enhance uterine contractility, an action also apparently antagonized by decidual relaxin.103,104
PLACENTAL TROPHOBLAST DEVELOPMENT AND VASCULARIZATION
During human and nonhuman primate pregnancy, the placenta simultaneously accesses the maternal blood and develops a vascular network for the transport of nutrients to and waste products from the fetus across the syncytiotrophoblast to ensure fetal growth and development. Both processes depend on the ability of the primordial stem-cell cytotrophoblasts to take either the villous pathway where they remain in the fetal compartment and differentiate morphologically into the syncytiotrophoblast or the extravillous pathway where they proliferate, aggregate into cell columns of the anchoring villi, and invade the endometrial stroma (Fig. 6).105 The syncytiotrophoblast covers the floating chorionic villi that become highly vascularized, whereas the extravillous cytotrophoblasts infiltrate the walls of the spiral arterioles to facilitate the process of placentation.
|

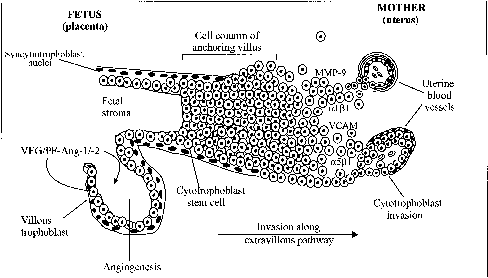
The vascular network within the placental villous core develops by in situ differentiation of fetal mesenchymal cells into vessels (vasculogenesis) and proliferation of existing vessels (angiogenesis) resulting in secondary and tertiary villi equipped with a functional arterio-capillary-venous system.34 Development of the uteroplacental circulation begins early in pregnancy with neovascularization of both fetal and maternal tissues.106,107,108 Neovascularization results in increased blood flow and increases in the effective exchange area crucial to support fetal development. In humans, placental vasculogenesis begins at approximately 21 days of gestation109 and continues through at least the 26th week of pregnancy.34 Angiogenesis accompanies vasculogenesis and is critical not only early in gestation but also important for early widespread extension of the fetal capillary system and later growth of the fetal vascular compartment during the final third of pregnancy.109,110 Several factors expressed by the placenta, including basic fibroblast growth factor,111,112,113 platelet-derived growth factor,114 placental growth factor,115,116 and vascular endothelial growth/permeability factor (VEG/PF)115 ,117 have been proposed as regulators of placental angiogenesis. Among these, VEG/PF selectively stimulates endothelial cell proliferation and the formation of new blood vessels.118,119 It also induces vascular permeability by actions on endothelial cells resulting in extravasation of plasma proteins that provide an extracellular fibrin matrix for angiogenesis.120 VEG/PF is encoded from a single gene and expressed as 5 isoforms having 121, 145, 165, 189, or 206 amino acids,121,122 the 121 and 165 isoforms exhibiting the greatest angiogenic activity.123,124,125 VEG/PF mRNA and protein are expressed by cytotrophoblasts, syncytiotrophoblast, and Hofbauer macrophage cells within the villous human placenta.114,117,124,126 Inactivation of the VEG/PF gene in transgenic mice results in significant defects in the vasculature of embryonic tissues and organs that are lethal.127
VEG/PF binds to two structurally related transmembrane tyrosine kinase receptors, VEG/PF flt-1 and KDR/flk-1,128,129 which are expressed on placental villous vascular endothelial cells126,129,130 and in villous and extravillous trophoblasts.117,126,131 Both receptors are essential for vascular development. Homozygous KDR/flk-1 defective mice die in utero as a result of an early defect in hematopoietic and endothelial cell development,132 and mouse embryos homozygous for the flt-1 mutation die because of failure to organize normal vascular channels.133
Two other closely related proteins, angiopoietin-1 and -2, work in concert with VEG/PF in signaling vascular morphogenesis by binding to the endothelial cell-specific transmembrane tyrosine kinase receptor Tie-2. In Tie-2 null mice, endothelial cells develop and assemble into tubes, but vessels are immature, lacking branching networks, encapsulation by periendothelial support cells, and proper organization into small and large vessels.134,135 These associations indicate that the Tie-2 receptor may also mediate the capacity of endothelial cells to recruit stromal cells that encase endothelial tubes for vessel stabilization. Transgenic/gene knock-out studies further indicate that angiopoietin-1 signals Tie-2 to recruit vascular support cells and that angiopoietin-2 inhibits this action136,137,138 by competitively inhibiting angiopoietin-1-induced kinase activation of the Tie-2 receptor. During early human pregnancy, angiopoietin-1 is localized to the cytotrophoblast and syncytiotrophoblast, angiopoietin-2 to the cytotrophoblast, and Tie-2 receptor to the endothelium, 139,140 observations that are consistent with the proposed role of the angiopoietin-Tie-2 system in development of the placental circulation.
Despite the importance of angiogenesis to neovascularization of the developing placenta, very little is known about the regulation of the process and the expression of the VEG/PF-angiopoitin-1/-2 system during human pregnancy. Although hypoxia is a potent stimulus of VEG/PF expression,141 estrogen has also been shown to regulate VEG/PF expression in the rat uterus142,143,144 and in human endometrial cells.145 Moreover, chronic estrogen treatment induced uterine angiogenesis in normal but not estrogen receptor-null transgenic mice.146 In the baboon, cytotrophoblast VEG/PF mRNA levels and vascularization of the villous placenta increase with advancing gestation in parallel with increasing placental estrogen production.147 Thus, the well-established role for estrogen in enhancing uteroplacental blood flow148, 149 may not only reflect changes in vascular reactivity but also enhanced angiogenesis.
At the same time the vascular system is developing within the chorionic villi, a select population of extravillous cytotrophoblasts migrate and invade the spiral arteries of the uterine endometrium at the placental-decidual junction (see Fig. 6). Histologic studies performed during the first half of human, baboon, and macaque pregnancy32, 150,151,152,153,154,155 demonstrate that cytotrophoblasts migrate to and colonize spiral arterioles/arteries by displacing endothelial cells from their basal lamina and partially or completely replacing the smooth muscle component within the tunica media. Consequently, the structure of spiral arteries and the dynamics of blood flow within them are modified by cytotrophoblast invasion, presumably to facilitate implantation and placentation.
As cytotrophoblasts differentiate into cells capable of invading the uterine stroma and blood vessels, their expression of adhesion molecules changes in the human,8,105,156,157,158,159 baboon,160 and rhesus monkey.154,161 As extravillous cytotrophoblasts migrate, expression of the integrin complex α5β1 and α6β4 is lost and that of the α1β1 laminin/collagen receptor is induced.158,162,163 Moreover, interaction of the α1β1 receptor with collagen type IV promotes, whereas interaction of the α5β1 receptor with fibronectin inhibits, invasion of human cytotrophoblasts in vitro.164,165 Zhou and associates105 have suggested that cytotrophoblasts balance invasion-retraining and invasion-promoting adhesion mechanisms as they differentiate. Migratory cytotrophoblast cells also express specific adhesion molecules, specifically vascular cell adhesion molecule (VCAM) and cadherins (e.g., VE-cadherin), that appear to secure cytotrophoblasts to each other and to the endothelium, thereby facilitating their migration against arterial blood flow.161,166,167 Interstitial and spiral arteriole invasion is also associated with the expression of matrix metalloproteinases (MMPs; e.g., MMP-9 collagenase), by intraluminal, extravasating, and intramural cytotrophoblasts,168,169 apparently to disrupt the extracellular matrices in the tunica media to allow cytotrophoblasts to modify the vessel wall. MMP-9 promotes cytotrophoblast invasion; the capacity of human cytotrophoblast to invade is completely inhibited by MMP-9 antibody in vitro.170 Clearly, the process of endovascular spiral artery invasion involves an intricately and temporally ordered expression of integrins, adhesion molecules, and proteinases by the extravillous cytotrophoblasts. This area of research is of intense interest and clinically relevant. Abnormal expression of several of these components has been observed in cytotrophoblasts of women who develop preeclampsia and in whom endovascular invasion is superficial.171 Unfortunately, our current understanding of the factors acting/interacting to regulate the timely expression of ECM and cell adhesion molecules by invading trophoblasts remains incomplete.